

Abstract
Background: Protein S (PS) is a 75-kDa, vitamin K-dependent anticoagulant that contains a Gla (enclosing γ-carboxyglutamic acids) module, a TSR (Thrombin Sensitive Region) module, four EGF (Epidermal Growth Factor)-like modules, and an SHBG (Sex Hormone Binding Globulin)-like region. Protein S deficiency is associated with risks for venous and arterial thrombosis, ischemic stroke, cerebral thrombophlebitis, myocardial infarction, and vascular calcification. Three disparate functions have been proposed for PS: (a) as a cofactor for active protein C; (b) as a cofactor of tissue factor pathway inhibitor, and (c) as a direct inhibitor of factor IXa. We have shown that PS binds FIXa with a Kd~40 nM. Currently, we are ascertaining the region and amino acid residues of FIXa that bind PS.
Results and Discussion: We demonstrated before that lysine residue K126 and K132 of FIXa were very important in binding PS. However, there were no information regarding the change in the conformation of the complex when these residues were mutated. Here, we determined the structural changes in the complex by circular dichroism (CD), isothermal titration calorimetry (ITC), mass spectrometry, and molecular modeling to define the basis of the FIXa-PS interaction.
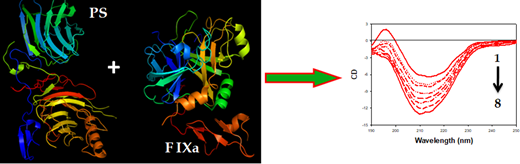
The circular dichroism (CD) titration data of interaction between PS and FIXa showed the conformational change of PS due to interaction with FIXa. In addition, to understand the stability of the complex, we determined the α-helicity changes of PS in the presence and absence of FIXa. Initially, PS exhibited only a small amount of α-helical content (~3.5%) with a peak maxima at 214 nm in CD spectra. On addition of a minute amount of FIXa (50 nM), the PS CD spectra were blue-shifted by 4 nm and, simultaneously, increased in α-helical content by ~12%. The massive change in PS α-helical content on binding to FIXa directly revealed the stability of the complex. A similar interaction pattern was reflected in ITC experiments. The binding of PS with FIXa was essentially entropy-driven; binding was exothermic and favored by negative enthalpy and large positive entropy changes. The large positive entropy contribution to the binding may result from the disruption of water networks surrounding both proteins and a condensation of sodium and calcium ions from the polypeptide chains. Also, the negative Gibbs free energy change in binding revealed the spontaneous association between the two proteins.
To achieve our goal of defining the amino acid interaction pattern of PS with FIXa, we combined liquid chromatography mass spectrometry (LCMS) and molecular modeling. To identify the residues involved in interaction, we subjected PS and FIXa, individually and in a complex, to a subtractive acetylation-protection assay. We used LCMS to compare the acetylated lysine residues in the individual proteins with the modified lysines in the complex." This methodology required normalization to lysine residues that were not acetylated. We found that lysine 126 (K126) and lysine 132 (K132) in FIXa were differentially protected in the complex about 2-fold compared with the free proteins. Our molecular modeling data also supported this mass spectrometric result. Modeling data pinpointed the regions of PS and FIXa that interact and revealed a major involvement of K132. To confirm the importance of K132, we analyzed by CD spectroscopy a derivative of FIXa in which K132 was changed to A132. We observed a dramatic reduction in interaction of the mutant FIXa with PS
Conclusion: Based on our preliminary data, we conclude that PS binds strongly with FIXa, and the complex is stabilized by a shift in α-helical content as shown in Figure 1.
Figure.1 shows the CD spectral change of PS (curve 1, right panel) due to addition of different concentration of FIXa (curve 2-8, right panel).
No relevant conflicts of interest to declare.

